The following analytical questions demonstrate how ActiveSite can be used to mine the data of our four black-footed ferret genomes and to begin thinking critically about the possibilities of research to pursue.
[vc_accordion collapsible=”yes” active_tab=”4″][vc_accordion_tab title=”How similar are the genomes of the domestic ferret and its relative the black-footed ferret?”]
This is an evolutionary question. The formula for answering this question is simple but not directly intuitive:

Not every SNP (mutation) identified in each black-footed ferret genome separates the entire black-footed ferret species from the domestic ferret species. The mutations that do separate these two species will have occurred long ago and have accumulated as the species evolved separately. A heterozygous mutation in a single individual is likely a newer mutation – important to population studies, but not to deep time evolution. A mutation that matters to deeper time will have become “fixed” within all black footed ferrets. These fixed differences are thus homozygous mutations possessed by all black-footed ferrets and are different from the domestic ferret. The number of fixed differences can be found on ActiveSite in the following way:

The total number of fixed differences is only part of the equation. To understand how much of the genome between the two species is the same, we need to know some information about the ferret genome. The genome used as the reference for this study is MusPutFur1.0, assembled by the Broad Institute. The info needed can be found on GenBank:
Image from: http://www.ncbi.nlm.nih.gov/assembly/GCA_000215625.1
The total size of the genome is not the number of base pairs being compared in ActiveSite; the number of base pairs being compared in ActiveSite is related to how much of the reference genome has actually been mapped by BFF DNA. By investigating ActiveSite we find that ~90% of the reference genome has been covered.
This means that the number of base pairs compared in ActiveSite is ~2.16 billion base pairs (2,400,000,000 × 0.9 = 2,160,000,000).
Now we can solve:
This number is only an approximation for several reasons:
- not every SNP identified is a single base pair; some are multiple base pairs and some are deleted base pairs, though the vast majority of SNPs do represent a single base pair difference
- only 90% of the full reference genome has been compared, meaning that 90% of the genomes of the two species are 99.6% similar
- four black-footed ferrets have been compared to only one domestic ferret genome, therefore the comparison is not even
These limitations in the data are not significant. With more samples and higher coverage the % similarity will not decrease or increase greatly from 99.6%. We can now begin estimating how long these two species have been evolving separately and investigate these fixed differences to analyze distinctions between the biology of the two unique species.
[/vc_accordion_tab][vc_accordion_tab title=”Which mutations are of most interest to conservation questions?”]
Of the 2.16 billion base pairs analyzed between all four black-footed ferrets a total of 19.5 million SNPs were found. This number is misleading though. As an industry standard, any SNP found is noted in the data – but some of these SNPs are the result of sequencing error and are not biologically real. Such sequencing error is rare and shows up in low frequency in the data. Given the majority of our data is called at a minimum of 8X coverage we have chosen to exclude SNPs that are below 25% frequency in the data (meaning that of eight reads at least two need to show the same SNP to be considered real). So first we must remove sequencing error:
We’re not done yet. For conservation science the SNPs that are of the most interest are found within the population – this means that the total fixed differences found in Question 1, which relate to evolution and are common to all four ferrets universally, are not informative to conservation.
This leaves us with ~1.3 million SNPs of high interest to conservation questions. These interesting SNPs encompass unique heterozygous and homozygous SNPs as well as heterozygous and homozygous SNPs shared by two or three ferrets, but not common to all four.
This number is a minimum constrained by how the coverage of each genome aligns to the coverage of other genomes (a SNP >0.24 frequency in one genome may not be >0.24 frequency in another, and thus it will be removed by the filters). When repeating the math with each individual ferret rather than the ferrets collectively, a range of 1.8-1.9 million SNPs are found that are potentially interesting to conservation research.
[/vc_accordion_tab][vc_accordion_tab title=”Which of the SDFZ cell lines would be of most value to restore to the captive breeding program?”]
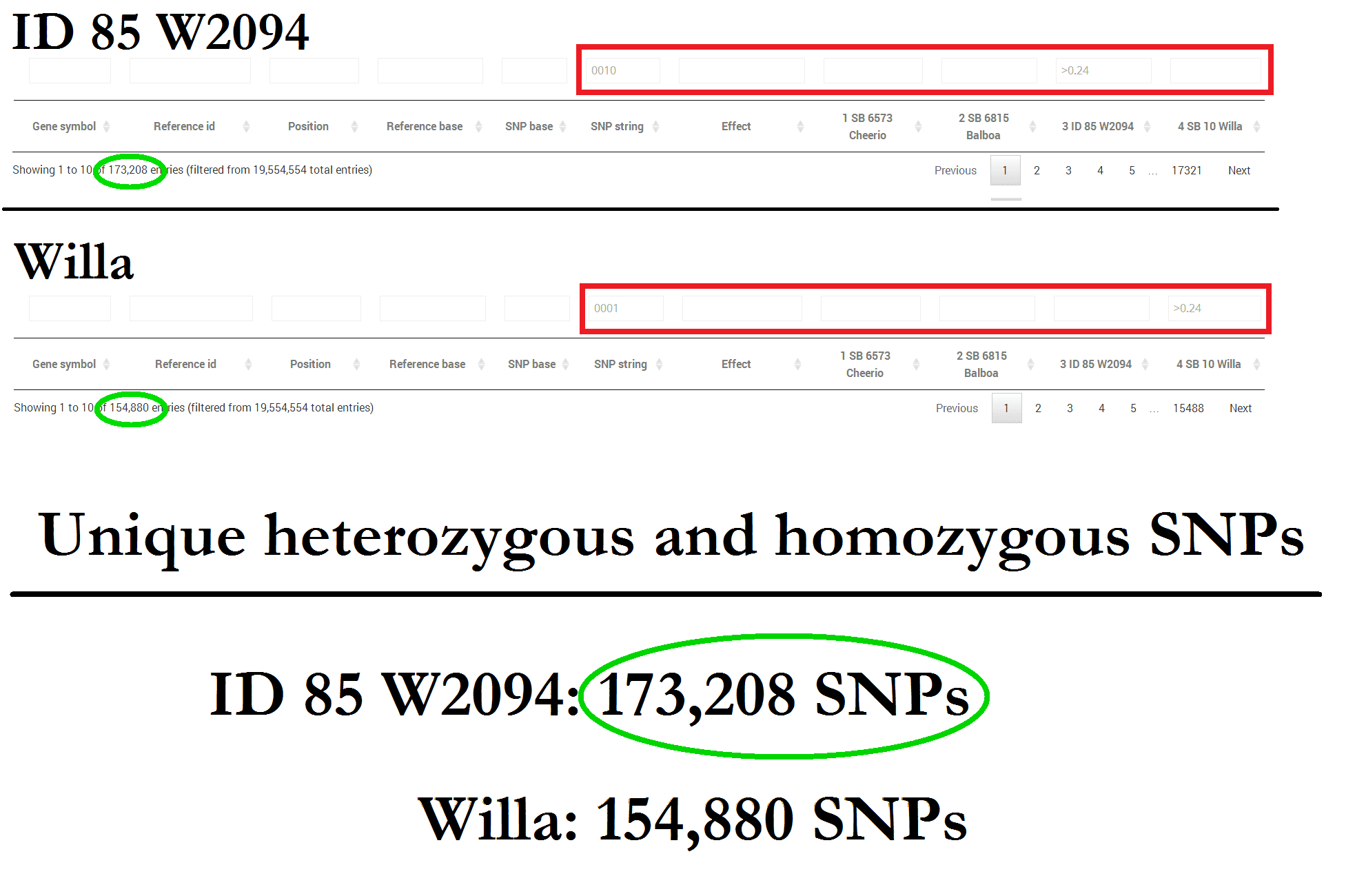
The goal of genetic rescue is to introduce new variation to the population. This means we need to know the total amount of unique mutations found in the genomes of the San Diego Frozen Zoo cell lines (Willa and ID 85 W2094).
The male ID 85 W2094 contains the highest amount of unique SNPs. A clone of ID 85 W2094 could be born and used to breed this unique genetic diversity into the population. Any of these mutations could be valuable to BFF fitness and survival.
[/vc_accordion_tab][vc_accordion_tab title=”What was the result of breeding two ferrets that lived 20 years apart through AI via cryopreserved sperm?”]
In our sample set the SDFZ frozen cell lines, while having no surviving descendents, were captured with the founders of the BFF breeding program at Meeteetse. Their genomes serve as a glimpse of the genetic structure of that Meeteetse remnant population and “generation 0” of the captive breeding program.
Cheerio represents a sample of the genetic structure of living ferrets – which all share descent from the founders of the captive breeding program. Due to inbreeding it has been assumed that genetic diversity, chiefly in levels of heterozygosity, has decreased from generation 0 to present living generations. This is found in our data.
Note the filter applied has changed. We are interested only in heterozygous SNPs per individual – meaning we apply the filter 0.25-0.75. Also, we want to identify the total mutations of each individual, so the SNP string uses underscores rather than zeros (the underscore allows a SNP to be present or absent in a ferret). In this way we identify the heterozygous SNPs found in an individual, regardless of whether they are unique or shared.
Balboa was the result of crossing 20-year-old sperm from generation 0 with a living black-footed ferret female. Therefore we expect his heterozygosity to be intermediate between the two generations – exactly halfway between the heterozygosity of living ferrets and generation 0. We can calculate an expected range for Balboa by finding the midpoints between the heterozygous SNP counts of Cheerio versus Willa and ID 85 W2094.
The answer is right in the range we expect it to be – telling us two important things:
- AI from cryopreserved sperm restores historic levels of genetic diversity exactly in the manner it is designed. By sequencing the genome of founder ferrets we can identify whether any of these restored SNPs have been completely lost in naturally breeding ferrets (like Cheerio).
- The two San Diego Frozen Zoo cell lines do adequately represent the levels of heterozygosity contained by the founders of the breeding program, both showing higher levels of diversity than Cheerio and Balboa, but placing Balboa appropriately in the middle range of our data sets observed heterozygosity counts. This means that as scientists continue to analyze more genomes, these numbers probably won’t change very much, and we will be able to start really deciphering what SNPs are the most significant to the conservation research of the black-footed ferret.
SNPs within the data tell a lot more about the individual ferrets than genome levels of heterozygosity. SNPs shared between two or three of the four BFF genomes can be identified to reveal kinship among the data set. SNPs that create differences in proteins and enzymes between two black-footed ferrets can be distinguished from silent mutations that do not create differences. Such different SNPs could be deleterious. Differences in these SNPs between early and late generations in the captive breeding program could be responsible for differences in phenotypes being observed. SNPs within genes related to the development of tissues, metabolism, reproduction, and immunity can also be explored.
While ActiveSite provides users unfamiliar with high throughput DNA data access to exploring genomes, these seemingly simple analytical questions can require intricate investigation. Further analyses beyond the examples outlined on this page require an understanding of the BFF individuals’ demography, an understanding of evolutionary genomics, and an introduction to bioinformatics data processing. A multitude of potential discoveries are possible through ActiveSite’s filters. While the answers to every question hinge on the limitations of the data, the questions that can be posed to the data are only limited by the scientific imagination of the inquirer.